Cystická fibróza (mukoviscidóza) – genetické onemocnění: příčiny, příznaky, léčba
Cystická fibróza (mukoviscidóza) – příčiny, příznaky a moderní léčba: genetika, diagnostika, péče a život s CF. Praktické informace pro pacienty a rodiny.
Cystická fibróza, známá také jako mukoviscidóza, CF a 65 růží, je onemocnění, které člověk může získat od svých rodičů. Způsobuje, že tělo vytváří hustý, lepkavý hlen, který se hromadí v plicích, trávicím systému a dalších částech těla.
Pokud mají oba rodiče gen pro cystickou fibrózu a předají ho svému dítěti, bude mít dítě cystickou fibrózu. Gen pro cystickou fibrózu musí mít každý z rodičů. Rodič nemusí mít cystickou fibrózu, ale přesto může mít gen. Osoba s cystickou fibrózou není nakažlivá (nemůže ji přenést na nikoho jiného). Cystická fibróza se nedá vyléčit, ale existuje mnoho léků, které pomáhají udržet lidi zdravé.
Galerie obrázků
4 Obrázky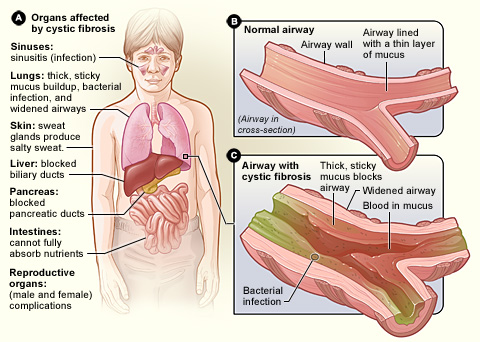

Příčiny a dědičnost
Cystická fibróza je způsobena mutacemi v genu CFTR, který řídí transport chloridových iontů přes buněčné membrány. Porucha tohoto kanálu vede k tvorbě hustého hlenu v různých orgánech. Onemocnění se dědí autosomálně recesivně — to znamená, že dítě onemocní tehdy, když zdědí vadnou kopii genu od obou rodičů. Pokud má jen jednu vadnou kopii, je člověk tzv. nosič (carrier) a obvykle má normální zdraví, ale může předat mutaci potomkům.
Příznaky
Příznaky se mohou lišit podle věku a závažnosti mutace, obvyklé jsou:
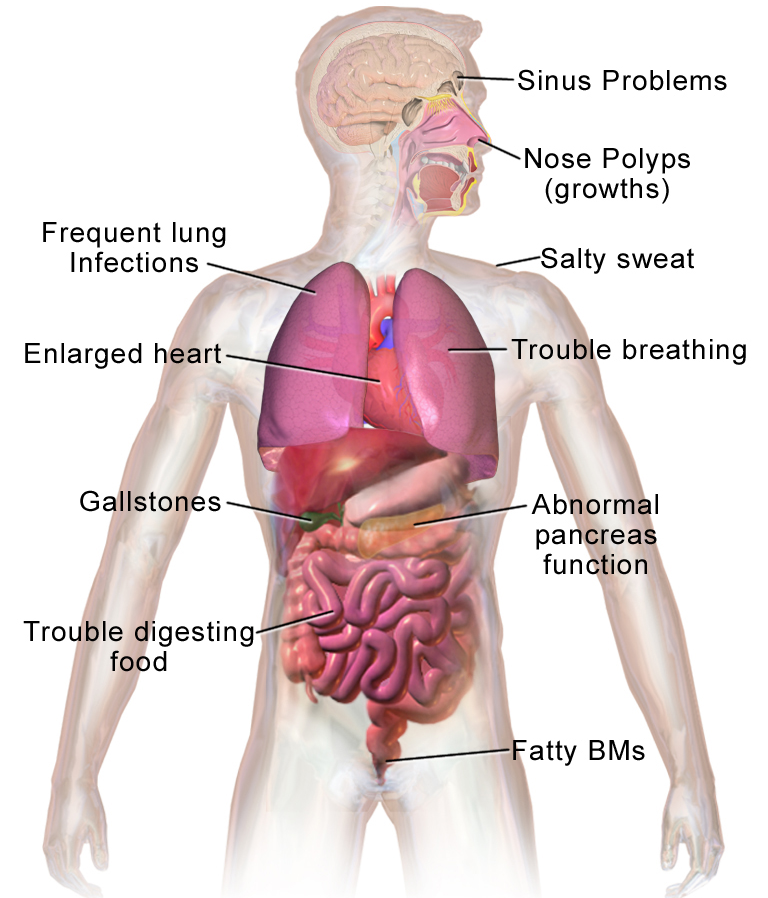
- Respirační symptomy: chronický kašel, nadměrné hleny, časté plicní infekce, dušnost, opakované záněty průdušek a vznik bronchiektázií.
- Trávicí obtíže: nedostatečná činnost slinivky (exokrinní pankreatická insuficience), mastná, páchnoucí stolice (steatorea), špatný přibývání na váze a poruchy vstřebávání tuků a vitamínů (A, D, E, K).
- Plodnost: u většiny mužů s CF je nepřítomnost chámovodu (CBAVD) a neplodnost, u žen může být snížená plodnost vlivem hustého hlenu.
- Další: časté solné ztráty u kojenců (riziko dehydratace a hypotonie), poruchy jater, diabetes související s CF (CFRD).
Diagnóza
- Novorozenecký screening: měření imunoreaktivního trypsinogenu (IRT) v krvi novorozenců — pozitivní screening vede k dalšímu vyšetření.
- Potní test (sweat test): zlatý standard pro stanovení zvýšené koncentrace chloridů v potu.
- Genetické testování: identifikace mutací v genu CFTR k potvrzení diagnózy a určení typu mutace (důležité pro cílenou léčbu).
- Funkční vyšetření plic: spirometrie a zobrazovací metody (RTG, CT plic) pro sledování postižení.
Léčba a komplexní péče
Cílem léčby je zlepšit funkci plic, předcházet a léčit infekce, zajistit výživu a kvalitu života. Péče je multidisciplinární a často probíhá v specializovaných centrech.
- Fyzioterapie dýchacích cest: techniky na odhlazování hlenu (perkusní masáž, dechová cvičení, PEP, autogenní drenáž).
- Inhalační léčba: bronchodilatancia, hypertonický roztok, mukolytika (např. dornáza alfa) pro zředění hlenu.
- Antibiotika: při akutních i chronických plicních infekcích — perorální, intravenózní nebo inhalované v závislosti na závažnosti a původci infekce (např. Pseudomonas aeruginosa).
- CFTR modulátory: moderní cílené léky, které vylepšují funkci proteinu CFTR u pacientů s určitými mutacemi (např. ivacaftor, kombinace lumacaftor/ivacaftor, tezacaftor/ivacaftor, elexacaftor/tezacaftor/ivacaftor). Tyto léky mohou významně zlepšit plicní funkce a kvalitu života u vhodných pacientů.
- Substituce pankreatických enzymů: u pacientů s pankreatickou insuficiencí k lepšímu trávení a vstřebávání živin; doplnění vitamínů rozpustných v tucích.
- Nutriční podpora: vysoce kalorická strava, doplňky, u některých pacientů sondová výživa.
- Imunizace a prevence: pravidelné očkování (např. proti chřipce, pneumokokům), hygienická opatření k prevenci přenosu infekcí mezi pacienty s CF.
- Transplantace plic: možnost u pokročilého postižení plic, když jiné terapie nestačí.
Genetické poradenství a prevence
Lidé, u kterých se v rodině vyskytuje CF nebo kteří jsou nosiči, by měli zvážit genetické vyšetření a poradenství. Možnosti zahrnují předporodní testování, prenatální diagnostiku a preimplantační genetickou diagnostiku (PGD) při asistované reprodukci. Poradenství pomáhá rodinám pochopit rizika a možnosti plánování rodiny.
Prognóza a kvalita života
Díky včasné diagnostice, lepší péči a novým léčebným možnostem se výrazně zlepšila délka a kvalita života lidí s CF. Průměrný věk dožití se v mnoha zemích posunul do dospělosti (často přes 40 let a více), ale prognóza závisí na závažnosti onemocnění, přístupu ke specializované péči a odpovědi na léčbu. Pravidelná péče ve specializovaném centru, adherence k léčbě a včasné řešení infekcí jsou klíčové.
Pokud máte podezření na cystickou fibrózu nebo jste nosičem mutace, poraďte se s praktickým lékařem, pediatrem nebo genetickým poradcem. Specializovaná centra pro CF poskytují komplexní péči a informace o nejnovějších léčebných možnostech.
Co CF dělá s tělem
Cystická fibróza postihuje celé tělo. Celkově má tělo problémy s přesunem soli do těch částí těla, které ji potřebují. Protože tělo má problémy s přesunem soli, hromadí se v místech, kde nemá, jako jsou plíce, žaludek a střeva.
Plíce
Když v plicích uvízne sůl, je v nich méně vody, což způsobuje, že hlen je velmi hustý. Velmi špatně se pak dýchá. Léčba tohoto stavu spočívá v podávání léků na dýchání, které pomáhají přidávat vodu do plic, aby hlen zůstal řidší a dal se lépe vykašlat. Když je hlen řidší a je ho méně, lépe se dýchá.
Léčba
Cystickou fibrózu nelze vyléčit. Přestože lidé mohou dělat věci, aby zůstali zdraví. Zdravé návyky brání tomu, aby člověk onemocněl ještě více. Lidé mohou zůstat čistí. Lidé se mohou držet dál od choroboplodných zárodků. Mohou pít vodu, která pomáhá odchodu hlenu. Užívání enzymů pomáhá trávit potravu, pokud je v žaludku hlen.
Cvičení odstraňuje hlen. Buduje silné svaly a kosti a posiluje plíce. Užívání vitaminů pomáhá tělu bojovat s infekcí. Pomáhá také tělu růst a dobře fungovat.
- Inhalační antibiotika se používají k tomu, aby se v hustém hlenu nerozmnožovaly bakterie.
- Inhalace slané vody pomáhá udržovat plíce hydratované.
Testování na cystickou fibrózu
- Test na chloridy v potu - testuje obsah soli v potu.
- Genetický test - používá se v případě pozitivního výsledku potního testu, aby se zjistilo, zda mají oba geny.
65 růží
"65 růží" je způsob, jakým některé děti označují svůj stav, protože cystická fibróza je pro malé dítě těžko vyslovitelná. Slovní spojení "65 růží" je také chráněno ochrannou známkou Nadace pro cystickou fibrózu, která pomáhá kontrolovat jeho používání. Pro malé děti je to velmi užitečný způsob porozumění. Při hlasitém vyslovení zní podobně jako cystická fibróza.
Otázky a odpovědi
Otázka: Co je to cystická fibróza?
Odpověď: Cystická fibróza je onemocnění, které způsobuje, že tělo produkuje hustý, lepkavý hlen, který se může hromadit v plicích, trávicím systému a dalších částech těla.
Otázka: Čím je cystická fibróza způsobena?
Odpověď: Cystická fibróza je způsobena zděděním genu pro cystickou fibrózu od obou rodičů.
Otázka: Může člověk, který má pouze jeden gen pro cystickou fibrózu, přenést toto onemocnění na své dítě?
Odpověď: Osoba, která má pouze jeden gen pro cystickou fibrózu, nemusí mít sama toto onemocnění, ale přesto může tento gen předat svému dítěti.
Otázka: Je cystická fibróza nakažlivá?
Odpověď: Ne, cystická fibróza není nakažlivá a nemůže se přenášet z jedné osoby na druhou.
Otázka: Lze cystickou fibrózu vyléčit?
Odpověď: Ne, v současné době neexistuje žádný lék na cystickou fibrózu, ale existuje mnoho léků, které mohou pomoci zvládnout toto onemocnění.
Otázka: Které části těla může cystická fibróza postihnout?
Odpověď: Cystická fibróza může postihnout plíce, trávicí systém a další části těla.
Otázka: Jak mohou lidé s cystickou fibrózou zůstat zdraví?
Odpověď: Lidé s cystickou fibrózou mohou zůstat zdraví, pokud užívají léky, sledují svůj stav a dodržují zdravý životní styl.
Související články
Autor
AlegsaOnline.com Cystická fibróza (mukoviscidóza) – genetické onemocnění: příčiny, příznaky, léčba Leandro Alegsa
URL: https://cs.alegsaonline.com/art/24952