Molekulární orbital: definice, principy a význam v chemii
Molekulární orbital: srozumitelná definice, principy a význam v chemii — kvantová mechanika, elektronová distribuce, předpovědi reaktivity a typy vazeb.
V chemii molekulový orbital (neboli MO) vysvětluje, co se děje s elektrony, když se atomy spojí v molekule. MO je matematická funkce, která popisuje vlnové chování elektronu v molekule. Chemici tyto funkce používají k předpovídání nebo vysvětlování chemických a fyzikálních vlastností. Funkce mohou například říci, jaká je pravděpodobnost, že se elektron nachází v nějaké konkrétní oblasti.
Chemici obvykle sestavují matematické modely molekulových orbitalů kombinací atomových orbitalů. Lze také použít hybridní orbitaly z každého atomu molekuly nebo jiné molekulové orbitaly ze skupin atomů. S těmito funkcemi mohou pracovat počítače. Molekulární orbitaly umožňují chemikům aplikovat kvantovou mechaniku při studiu molekul. MO odpovídají na otázky, jak atomy v molekulách drží pohromadě. Různé zaoblené tvary v orbitálním diagramu označují, kde by se v atomu nejspíše nacházely elektrony.
Galerie obrázků
5 Obrázky
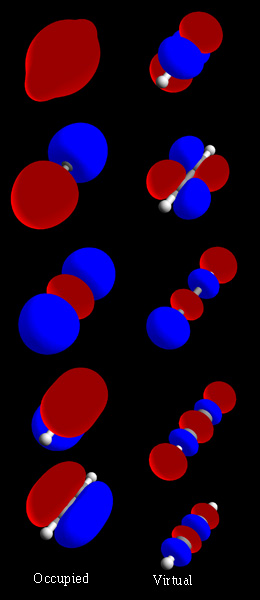



Základní princip: LCAO a superpozice
Jednoduchý způsob, jak vznikají molekulové orbitaly, je metoda LCAO (linear combination of atomic orbitals) – lineární kombinace atomových orbitalů. Když se dva atomové orbitaly navzájem kombinují, vznikají dva výsledné molekulární orbitaly:
- vazebný orbital (bonding) – amplitudy vlnových funkcí mají stejný smysl, dochází k zvětšení elektronové hustoty mezi jádry a k energetickému snížení;
- protivazebný orbital (antibonding) – amplitudy mají opačný smysl, mezi jádry vzniká uzel a orbital má vyšší energii.
Kromě toho může vzniknout neutrální (nonbonding) orbital, jehož energie a tvar jsou podobné původnímu atomovému orbitalu a který výrazně neovlivňuje vazbu.
Typy molekulových orbitalů
- Sigma (σ) – vznikají podél osy spojující jádra; často z 1s, sp nebo pz orbitalů; mají kruhovou symetrii kolem osy vazby.
- Pi (π) – vznikají z bočního překrytí p orbitalů; mají uzly na ose mezi jádry a charakteristickou dvoulobou symetrii.
- Delta (δ) – raritnější, vyskytují se u některých přechodných kovů (překrytí d orbitalů).
Obsazování orbitalů a pravidla
Elektrony v molekulových orbitalech se řídí stejnými kvantovými pravidly jako v atomech:
- Paulího vylučovací princip – v jednom orbitalu mohou být maximálně dva elektrony s opačnými spiny.
- Hundovo pravidlo – u orbitalů stejné energie se elektrony obsazují s paralelními spiny, pokud je to možné.
- Pořadí energií orbitalů závisí na molekule; pro jednoduché diatomické molekuly je často ukazováno v MO diagramech (např. pro H2, He2, Li2, ...).
Vazebný řád lze odhadnout podle počtu elektronů v vazebných (nbonding) a protivazebných (nantibonding) orbitalech pomocí výrazu:
vazebný řád = (nbonding − nantibonding) / 2
HOMO, LUMO a reaktivita
Nejvyšší obsazený molekulový orbital se označuje HOMO (Highest Occupied Molecular Orbital) a nejnižší volný orbital LUMO (Lowest Unoccupied Molecular Orbital). Energie a složení HOMO a LUMO hrají klíčovou roli v chemické reaktivitě, fotochemii a spektru molekuly. Malý rozdíl energie HOMO–LUMO obvykle znamená vyšší reaktivitu a absorpci světla při delším vlnovém délkovém rozsahu (nižší energetické excitace).
Příklady a důsledky v praxi
- H2: jednoduchý případ, kde kombinace dvou 1s orbitalů dává vazebný σ1s a protivazebný σ*1s; obsazením dvou elektronů v σ1s vzniká stabilní jednostranná vazba.
- O2: má v MO diagramu dva nespárované elektrony v π* (antibonding) orbitalech, což vysvětluje paramagnetické chování kyslíku.
- N2: vysoký vazebný řád (trojná vazba) – pevná a krátká vazba, malé reaktivita vůči běžným činidlům.
- Benzén: delokalizované π orbitaly nad a pod rovinou kruhu vedou k aromatické stabilizaci a specifickým spektrálním vlastnostem.
Výpočtové metody a interpretace
Popis MO se provádí analyticky i numericky. Mezi běžné výpočetní metody patří:
- Hartree–Fock (HF) – základní kvantově-chemická metoda, která používá jednoelektronové orbitaly a průměrný elektrostatický efekt elektronů;
- DFT (density functional theory) – velmi používaná metoda pro výpočty elektronové hustoty a energetických vlastností s dobrým poměrem přesnost / výpočetní náročnost;
- post‑Hartree–Fock (MP2, CI, CCSD(T) apod.) – zahrnují korelace elektronů a poskytují vyšší přesnost za cenu vyšší výpočetní náročnosti.
Počítače a vizualizační programy umožňují kreslit tvary MO, izoplochy elektronové hustoty a energetické diagramy. Tyto obrazy pomáhají pochopit, kde se elektron nachází a jak orbitaly ovlivňují vlastnosti molekuly.
Význam v chemii a aplikace
Molekulární orbitaly jsou zásadní pro pochopení a predikci:
- typů a síly chemických vazeb,
- magnetických vlastností (paramagnetismus vs. diamagnetismus),
- spektrálních přechodů (UV/VIS, fotochemie),
- reaktivity a mechanizmů organických reakcí (fronty HOMO/LUMO),
- materiálových vlastností, katalýzy a navrhování nových molekul v chemii a farmacii.
Tipy pro studenty
- Začněte jednoduchými příklady (H2, He2, Li2, Be2, B2, C2, N2, O2, F2) a sledujte, jak se mění MO diagram při zvyšujícím se počtu elektronů.
- Procvičujte výpočet vazebného řádu a určování paramagnetismu podle obsazení π* orbitalů.
- Využívejte vizualizační nástroje k zobrazení tvarů orbitalů – obraz často pomůže pochopit abstraktní matematiku.
Celkově jsou molekulární orbitaly mocným rámcem, který propojuje kvantovou mechaniku s pozorovatelnými chemickými vlastnostmi a poskytuje praktické nástroje pro výzkum i výuku.

Historie
Slovo orbitální poprvé v angličtině použil Robert S. Mulliken. O MO psal již dříve německý fyzik Erwin Schrödinger. Schrödinger je nazýval Eigenfunktion.
Fyzik Max Born popsal teorii molekulových orbitalů v roce 1926. Dnes je známá jako Bornovo pravidlo a je součástí kodaňské interpretace kvantové mechaniky. Když byla tato teorie původně navržena, nesouhlasila s modelem atomu Nielse Bohra. Bohrův model popisoval elektrony jako "obíhající" kolem jádra, jak se pohybují v kruzích. Bornův model si však nakonec získal oblibu, protože dokázal popsat umístění elektronů v molekulách a vysvětlil řadu dříve nevysvětlitelných chemických reakcí.
Přehled
Atomové orbitaly předpovídají polohu elektronu v atomu. Molekulární orbitaly vznikají spojením atomových orbitalů. Molekulový orbital může poskytnout informace o elektronové konfiguraci molekuly. Elektronová konfigurace udává nejpravděpodobnější polohu a energii jednoho (nebo jednoho páru) elektronu (elektronů). Většinou se MO znázorňuje jako lineární kombinace atomových orbitalů (metoda LCAO-MO), zejména při přibližném použití. To znamená, že chemici předpokládají, že pravděpodobnost výskytu elektronu v libovolném místě molekuly je součtem pravděpodobností výskytu elektronu v tomto místě na základě jednotlivých atomových orbitalů. LCAO-MO je jednoduchý model vazby v molekulách a je důležitý pro studium teorie molekulových orbitalů.
Teoretičtí chemici používají počítače k výpočtu MO různých molekul (reálných i imaginárních). Počítač dokáže nakreslit grafy "mraku", které ukazují, s jakou pravděpodobností se elektron bude nacházet v libovolné oblasti. Počítače mohou také poskytnout informace o fyzikálních vlastnostech molekuly. Mohou také říci, kolik energie je potřeba k vytvoření molekuly. To pomáhá chemikům říci, zda lze některé malé molekuly spojit do větších molekul.
Většina současných způsobů výpočetní chemie začíná výpočtem MO systému. Elektrické pole každého MO je generováno jádry všech atomů a určitým průměrným rozložením ostatních elektronů.
Analogie
Pochopit MO je jako zjistit, kde se ve velkém obchodě s domácími potřebami nachází který zaměstnanec (aniž byste se podívali dovnitř obchodu). Analytik zná počet zaměstnanců pracujících v obchodě a oddělení každého zaměstnance. Ví také, že si zaměstnanci navzájem nešlapou na paty a že zaměstnanci stojí spíše v uličce než na regálech se zbožím. Zaměstnanci opouštějí své vlastní oddělení, aby pomohli zákazníkům najít zboží v jiných odděleních nebo zkontrolovat zásoby. Analytik udávající polohu všech zaměstnanců v prodejně ve vybraném okamžiku, aniž by se podíval dovnitř, je jako chemik, který vypočítává MO molekuly. Stejně jako MO nelze určit přesnou polohu každého elektronu, není známa ani přesná poloha každého zaměstnance. MO, který má uzlovou rovinu, je jako závěr, že zaměstnanci chodí uličkami, a ne regály. Přestože elektrony přispívají z určitého atomu, elektron vyplňuje MO bez ohledu na jeho zdrojový atom. Je to podobné, jako když zaměstnanec během dne opustí své oddělení a prochází se jinde v obchodě. MO je tedy neúplným popisem elektronu, stejně jako jsou výpočty analytika o neviditelném obchodě neúplným odhadem umístění zaměstnanců.

Tvorba molekulových orbitalů
Teoretičtí chemici vynalezli pravidla pro výpočet MO. Tato pravidla vycházejí z pochopení kvantové mechaniky. Kvantová mechanika pomáhá chemikům využít fyzikální poznatky o elektronech k tomu, aby zjistili, jak se elektrony chovají v molekulách. Molekulové orbitaly vznikají z "povolených" interakcí mezi atomovými orbitaly. (Interakce jsou "povolené", pokud jsou symetrie (určené z teorie grup) atomových orbitalů vzájemně kompatibilní.) Chemici studují interakce atomových orbitalů. Tyto interakce vycházejí z překryvu (míra toho, jak dobře spolu dva orbitaly konstruktivně interagují) mezi dvěma atomovými orbitaly. Překrytí je důležité, pokud jsou si atomové orbitaly energeticky blízké. A konečně, počet MO v molekule se musí rovnat počtu atomových orbitalů v atomech, které se spojují, aby vytvořily molekulu.
Kvalitativní přístup
Aby mohli chemici diskutovat o struktuře molekul, musí rozumět geometrii MO. Metoda LCMO (lineární kombinace atomových orbitalů molekulových orbitalů) poskytuje hrubý, ale dobrý popis MO. V této metodě jsou molekulové orbitaly vyjádřeny jako lineární kombinace všech atomových orbitalů každého atomu v molekule.
Lineární kombinace atomových orbitalů (LCAO)
Molekulární orbitaly poprvé představili Friedrich Hund a Robert S. Mulliken v letech 1927 a 1928.
Aproximaci lineární kombinace atomových orbitalů neboli "LCAO" pro molekulové orbitaly zavedl v roce 1929 Sir John Lennard-Jones. Jeho průlomová práce ukázala, jak z kvantových principů odvodit elektronickou strukturu molekul fluoru a kyslíku. Tento kvalitativní přístup k teorii molekulových orbitalů je součástí počátku moderní kvantové chemie.
Lineární kombinace atomových orbitalů (LCAO) lze použít k odhadu molekulových orbitalů, které vznikají při vazbě atomů molekuly. Podobně jako pro atomový orbital lze i pro molekulový orbital sestavit Schrodingerovu rovnici, která popisuje chování elektronu. Lineární kombinace atomových orbitalů (součty a rozdíly atomových vlnových funkcí) poskytují přibližná řešení molekulových Schrödingerových rovnic. Pro jednoduché dvouatomové molekuly jsou vlnové funkce, které získáte, matematicky reprezentovány rovnicemi
Ψ = c ψ aa+ c ψ bb
a
Ψ* = c ψ aa- c ψ bb
kde Ψ a Ψ* jsou molekulové vlnové funkce pro vazebné a antivazebné molekulové orbitaly, ψa a ψb jsou atomové vlnové funkce z atomů a a b a ca a c bjsou nastavitelné koeficienty. Tyto koeficienty mohou být kladné nebo záporné v závislosti na energiích a symetriích jednotlivých atomových orbitalů. Jak se dva atomy přibližují k sobě, jejich atomové orbitaly se překrývají a vytvářejí oblasti s vysokou elektronovou hustotou. Mezi oběma atomy tak vznikají molekulové orbitaly. Atomy drží pohromadě elektrostatická přitažlivost mezi kladně nabitými jádry a záporně nabitými elektrony, které obsazují vazebné molekulové orbitaly.
Vazebné, nevazebné a nevazebné MOs
Při interakci atomových orbitalů mohou vznikat tři typy molekulových orbitalů: vazebné, antivazebné a nevazebné.
MO pro lepení:
- Vazbové interakce mezi atomovými orbitaly jsou konstruktivní (fázové) interakce.
- Vazbové MO mají nižší energii než atomové orbitaly, které je vytvářejí.
Protispojivé MO:
- Antivazbové interakce mezi atomovými orbitaly jsou destruktivní (mimofázové) interakce.
- Antivaze MO mají vyšší energii než atomové orbitaly, které je vytvářejí.
Nevázané MO:
- Nevazebné MO jsou výsledkem toho, že mezi atomovými orbitaly neprobíhá žádná interakce kvůli nedostatku kompatibilních symetrií.
- Nevazebné MO budou mít stejnou energii jako atomové orbitaly jednoho z atomů v molekule.
HOMO a LUMO
Každý molekulový orbital má svou vlastní energetickou hladinu. Chemici řadí MO podle energetických hladin. Chemici předpokládají, že elektrony nejprve zaplní MO s nejnižší energetickou hladinou. Například pokud má molekula elektrony k zaplnění 15 orbitalů, bude zaplněno 15 MO s nejnižší energetickou hladinou. Patnáctý MO na seznamu by se nazýval "nejvýše obsazený molekulový orbital" (HOMO) a šestnáctý MO na seznamu by byl "nejnižší neobsazený molekulový orbital" (LUMO). Rozdíl mezi energetickou hladinou HOMO a energetickou hladinou LUMO se nazývá pásová mezera. Pásová mezera může někdy sloužit jako měřítko excitability molekuly: čím menší je její energie, tím snadněji se excituje. Když je elektron excitován, přeskočí na neobsazený MO. To může například pomoci odhadnout, zda něco bude vydávat světlo (luminiscence).

Otázky a odpovědi
Otázka: Co je to molekulární orbital?
Odpověď: Molekulární orbital (neboli MO) je matematická funkce, která popisuje vlnové chování elektronu v molekule. Vysvětluje, co se děje s elektrony, když se atomy spojí v molekule, a může říci, jaká je pravděpodobnost, že se elektron nachází v určité oblasti.
Otázka: Jak chemici sestavují matematické modely molekulových orbitalů?
Odpověď: Chemici obvykle sestavují matematické modely molekulových orbitalů kombinací atomových orbitalů. Lze také použít hybridní orbitaly z každého atomu molekuly nebo jiné molekulové orbitaly ze skupin atomů. S těmito funkcemi mohou pracovat počítače.
Otázka: Co má kvantová mechanika společného se studiem molekul?
Odpověď: Molekulární orbitaly umožňují chemikům použít kvantovou mechaniku ke studiu molekul. Odpovídají na otázky o tom, jak atomy v molekulách drží pohromadě, a umožňují nahlédnout do chemických a fyzikálních vlastností.
Otázka: Co jsou to orbitální diagramy?
Odpověď: Orbitální diagramy jsou vizuální znázornění, která ukazují, kde by se elektrony v atomu s největší pravděpodobností nacházely na základě jeho různých zaoblených tvarů.
Otázka: Jak fungují hybridní orbitaly?
Odpověď: Hybridní orbitaly kombinují různé typy atomových orbitalů do jednoho nového typu, který má jedinečné vlastnosti ve srovnání se svými složkami. Tyto hybridy se často používají při vytváření matematických modelů molekulových orbitalů.
Otázka: Jak mohou počítače pomoci při studiu MO?
Odpověď: Počítače mohou pomoci při studiu MO tím, že pracují na jejich funkcích a poskytují přesnější předpovědi nebo vysvětlení chemických a fyzikálních vlastností v molekulách.
Související články
Autor
AlegsaOnline.com Molekulární orbital: definice, principy a význam v chemii Leandro Alegsa
URL: https://cs.alegsaonline.com/art/65861
Zdroje
- nobelprize.org : Nobelprize.org