Amyotrofická laterální skleróza (ALS) – příznaky, příčiny a léčba
ALS (amyotrofická laterální skleróza): zjistěte příznaky, příčiny a možnosti léčby, péči a podporu pro pacienty a rodiny.
Nemoc motorického nervu (někdy nazývaná Lou Gehrigova choroba nebo amyotrofická laterální skleróza) je chronické, progresivní a v drtivé většině případů smrtelné neurologické onemocnění. Postihuje motorické neurony — nervové buňky, které přenášejí pokyny z mozku a míchy do svalů, takže tyto svaly mohou pracovat.
V ALS postupně odumírají horní i dolní motorické neurony. To vede ke svalové slabosti, úbytku svalové hmoty (atrofii) a k postupné ztrátě schopnosti provádět dobrovolné pohyby, například mluvit, chodit nebo polykat. Nemoc se časem zhoršuje a způsobuje rostoucí invaliditu a nakonec i smrt, obvykle v důsledku selhání dýchacího svalstva. Neexistuje zatím univerzální lék, ale existují léčebné postupy, které mohou zpomalit průběh u některých pacientů a zlepšit kvalitu života.
Amyotrofická laterální skleróza (ALS) patří mezi pět hlavních typů onemocnění motorického neuronu a je nejčastější z nich. Přibližně 5 až 10 % případů bývá zděděno po rodičích (rodinná ALS); většina případů je sporadická. S průběhem nemoci může být spojena i změna chování nebo porucha řeči a jazyka; u části pacientů se rozvine frontotemporální demence.
Galerie obrázků
10 Obrázky





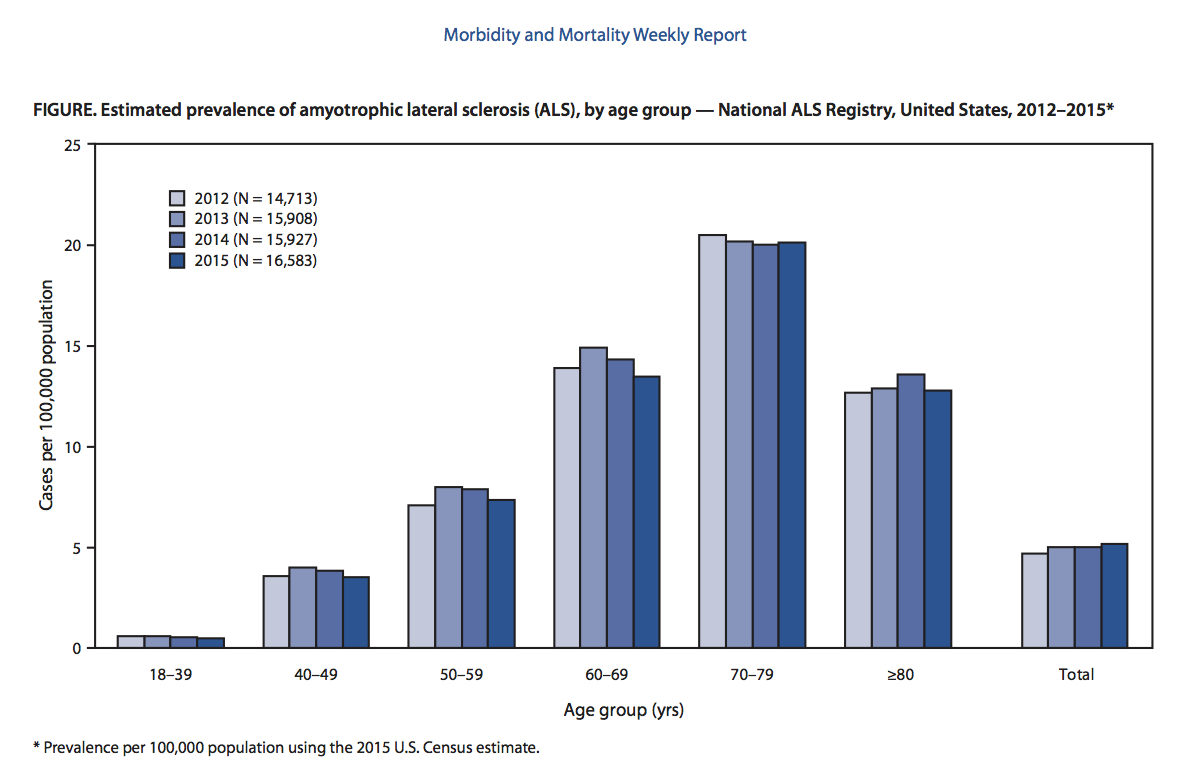



Příznaky
Počáteční příznaky mohou být nenápadné a liší se podle toho, které svaly jsou postiženy jako první. Mezi časté příznaky patří:
- oslabení nebo ztuhlost svalů končetin (např. potíže se zvedáním paže, chůzí),
- záškuby svalů (fascikulace) a křeče,
- obtíže s mluvením (dysartrie) a polykáním (dysfagie),
- úbytek svalové hmoty (svalová atrofie),
- poruchy dýchání v pozdějších stádiích (dušnost zejména při ležení),
- u některých pacientů změny v chování, myšlení nebo jazyce.
Příčiny a rizikové faktory
Přesná příčina ALS není plně objasněna. Onemocnění je spojeno s kombinací genetických a environmentálních faktorů. Mezi známé aspekty patří:
- Genetika: U části pacientů jsou identifikovatelné genetické mutace (např. mutace v genu SOD1 nebo v genu C9orf72) — proto se v rodinných případech doporučuje genetické poradenství.
- Věk a pohlaví: Nejčastěji postihuje dospělé kolem 50–70 let; mírně častěji muže než ženy.
- Možné rizikové faktory: kouření, některé expozice toxickým látkám nebo opakované poranění nervového systému byly navrženy, ale přímý příčinný vztah není definitivně prokázán.
Diagnostika
Diagnóza ALS je založena na klinickém vyšetření a elektromyografii (EMG), která prokazuje poškození motorických neuronů. Další vyšetření slouží k vyloučení jiných příčin příznaků:
- MRI mozku a páteře pro vyloučení strukturálních onemocnění,
- elektromyografie (EMG) a vyšetření rychlosti vedení nervů,
- laboratorní krevní testy k vyloučení metabolických nebo autoimunitních příčin,
- genetické testování v případě podezření na familiární formu nebo na základě rodinné anamnézy.
Léčba a péče
Aby se maximalizovala kvalita života a funkční schopnosti, je léčba u ALS obvykle multidisciplinární a zahrnuje specializované týmy (neurolog, fyzioterapeut, ergoterapeut, logoped, nutriční specialista, plicní lékař a odborník na paliativní péči).
- Farmakologická léčba: existují léky, které mohou zpomalit progresi u některých pacientů — nejznáměji riluzol, který mírně prodlužuje přežití, a edaravone (v některých zemích dostupný), které může zpomalit pokles funkce u vybraných pacientů. V posledních letech se vyvíjejí a v některých případech schvalují i nové cílené terapie zaměřené na konkrétní genetické mutace.
- Podpora dýchání: neinvazivní ventilace (noční kontinuální pozitivní tlak nebo NIV) může výrazně zlepšit dýchání, spánek a kvalitu života; v pokročilých stádiích může být zvažována tracheostomie s dlouhodobou ventilací.
- Výživa: poruchy polykání se řeší úpravou stravy a v případě potřeby zavedením enterální výživy (PEG), aby se předešlo podvýživě a riziku aspirace.
- Fyzikální a logopedická rehabilitace: pomáhají udržet pohyblivost, předcházet ztuhlosti, zlepšovat komunikaci a polykání.
- Paliativní péče a psychologická podpora: zaměřena na zmírnění příznaků, plánování péče a podporu rodin a pečovatelů. Důležitá je také péče o duševní zdraví pacienta a rodiny.
Průběh a prognóza
Průběh ALS je velmi variabilní. Někteří pacienti mohou mít rychlý pokles funkce během měsíců, jiní žijí mnoho let s pomaleji progredující chorobou. Průměrná délka přežití od stanovení diagnózy bývá 3–5 let, ale u části pacientů je delší. Kvalitu života může významně ovlivnit včasná odborná péče a podpora.
Život s ALS a podpora
Život s ALS vyžaduje přizpůsobení domácnosti, technickou asistenci (pomůcky pro pohyb, komunikaci, domácí ventilaci) a sociální podporu. Multidisciplinární centra pro ALS pomáhají koordinovat péči a nabízejí poradenství pro pacienty i jejich rodiny. Důležité je také plánování před lékařskými rozhodnutími (např. ohledně ventilace nebo výživy) a kontakt na pacientské organizace a podpůrné skupiny.
Kdy vyhledat lékaře
Pokud se objeví postupné zeslabení svalstva, záškuby svalů, potíže s polykáním nebo mluvením, je vhodné vyhledat neurologické vyšetření co nejdříve. Včasná diagnostika umožňuje rychleji zahájit podpůrná opatření a zapojit multidisciplinární péči.
Prevence: Specifická prevence ALS není známa. U rodinných případů je možné genetické poradenství. Výzkum pokračuje v hledání rizikových faktorů a nových léčebných přístupů.
Navzdory ztrátě kontroly nad svalovými funkcemi si mnozí pacienti uchovávají myšlení a paměť; u části pacientů však může dojít ke změnám v osobnosti a kognitivních funkcích — někteří pacienti rozvíjejí poruchy, které ovlivní inteligenci, paměť nebo osobnost, jakou měli před nemocí. Včasná podpora a vhodná péče jsou klíčové pro udržení co nejlepší kvality života.
Příznaky
Onemocnění motorického neuronu nevykazuje mnoho příznaků, a proto je velmi obtížné ho diagnostikovat. Obvykle postihuje lidi ve věku 40-60 let. Mezi nejčasnější příznaky mohou patřit záškuby, křeče nebo ztuhlost svalů, svalová slabost postihující ruku nebo nohu, nezřetelná a podivně znějící nosová řeč nebo obtížné žvýkání či polykání. Tyto obecné příznaky pak přerostou ve zřetelnou slabost nebo atrofii, která může lékaře přivést k domněnce, že člověk trpí ALS.
Části těla postižené časnými příznaky ALS závisí na tom, které svaly v těle jsou postiženy jako první. Přibližně 75 % lidí má ALS s nástupem končetin. V některých z těchto případů příznaky nejprve postihnou jednu z nohou a pacienti mají neobratnost při chůzi nebo běhu nebo si všimnou, že častěji zakopávají nebo se potácejí. U jiných pacientů se projevy nemoci nejprve projeví na ruce nebo paži, protože mají potíže s jednoduchými úkony vyžadujícími obratnost rukou nebo schopnost pohybovat drobnými věcmi, jako je zapínání knoflíků na košili, psaní nebo otáčení klíčem v zámku.
Přibližně 25 % případů tvoří ALS s bulbárním začátkem. Tito pacienti mají nejprve potíže s jasnou řečí. Řeč se stává obtížně srozumitelnou a nezřetelnou. Mluvení nosem a tišší mluvení jsou často prvními příznaky. Následují potíže s polykáním a ztráta pohyblivosti jazyka. Nakonec dochází k úplné ztrátě řeči a schopnosti udržet volné dýchací cesty při polykání.
Léčba
Na ALS neexistuje žádný lék. Úřad pro kontrolu potravin a léčiv (FDA) však schválil první lék na toto onemocnění: riluzol. Předpokládá se, že riluzol snižuje poškození motorických neuronů. Klinické studie s pacienty s ALS ukázaly, že riluzol dává lepší šanci na přežití o několik měsíců, a to hlavně u těch, kteří mají potíže s polykáním. Lék také umožňuje delší čas, než pacient bude potřebovat pomoc při dýchání pomocí ventilátoru nebo tracheotomie. Riluzol neléčí již vzniklé poškození motorických neuronů.
Další léčba ALS má za cíl zmírnit bolestivé příznaky a zlepšit kvalitu života pacientů.
Otázky a odpovědi
Otázka: Co je to onemocnění motorického neuronu?
Odpověď: Onemocnění motorického neuronu je chronické, progresivní a smrtelné neurologické onemocnění, které způsobuje odumírání nervových buněk v centrální nervové soustavě, což vede k narůstající invaliditě a nakonec ke smrti.
Otázka: Co dělají nervové buňky postižené onemocněním motorického neuronu?
Odpověď: Nervové buňky postižené onemocněním motorického neuronu ovládají dobrovolné svalové pohyby, jako je mluvení, chůze, polykání a pohyb těla.
Otázka: Lze onemocnění motorického neuronu vyléčit?
Odpověď: Ne, na onemocnění motorického neuronu neexistuje žádný známý lék.
Otázka: Co je amyotrofická laterální skleróza (ALS)?
Odpověď: Amyotrofická laterální skleróza (ALS) je nejčastějším typem onemocnění motorického neuronu, které způsobuje svalovou slabost a zmenšování svalů v celém těle, což vede ke svalové atrofii.
Otázka: Co způsobuje svalovou atrofii u ALS?
Odpověď: Odumírají horní i dolní motorické neurony, takže přestávají vysílat zprávy do svalů, což vede ke svalové atrofii.
Otázka: Může být onemocnění motorického neuronu dědičné?
Odpověď: Ano, asi 5 až 10 % případů onemocnění motorického neuronu se dědí přímo po rodičích.
Otázka: Má onemocnění motorického neuronu vliv na inteligenci, paměť a osobnost člověka?
Odpověď: I pacienti v pozdějších stádiích onemocnění motorického neuronu mohou mít stále stejnou inteligenci, paměť a osobnost jako před začátkem onemocnění.
Související články
Autor
AlegsaOnline.com Amyotrofická laterální skleróza (ALS) – příznaky, příčiny a léčba Leandro Alegsa
URL: https://cs.alegsaonline.com/art/66919
Zdroje
- ninds.nih.gov : "Motor neuron diseases fact sheet: National Institute of Neurological Disorders and Stroke (NINDS)"