Osteogenesis imperfecta – dědičná nemoc křehkých kostí: příčiny, příznaky a typy
Osteogenesis imperfecta – dědičná nemoc křehkých kostí: zjistěte příčiny, příznaky a typy, rizika zlomenin a ztráty sluchu. Praktický přehled pro pacienty a rodiny.
Osteogenesis imperfecta je genetická porucha, známá také jako nemoc křehkých kostí. Nejčastěji se jedná o autozomálně dominantní onemocnění — to znamená, že nemoc se může projevit, pokud je přítomen patologický gen od jediného rodiče — nicméně byly popsány i autosomálně recesivní a de novo mutace. Onemocnění postihuje především strukturu kolagenu typu I, tedy část kostní tkáně označovanou v článku jako kolagenní tyčinka, která zajišťuje pevnost kostí. Patologie vede k oslabení nebo chybné tvorbě kolagenních vláken, což způsobuje křehkost kostí. Poprvé byl tento stav popsán Vrolikem v roce 1854.
Galerie obrázků
10 Obrázky







Příčiny
Nejčastější příčinou jsou mutace v genech COL1A1 a COL1A2, které kódují řetězce kolagenu typu I. Tyto mutace mohou:
- způsobit snížené množství normálního kolagenu (typicky mírnější forma),
- nebo vytvářet abnormální kolagen, který naruší strukturu kostí (častěji těžší formy).
Kromě uvedených genů byly identifikovány i další genetické odchylky odpovědné za vzácnější formy OI. V některých případech vzniká mutace nově u postiženého jedince (de novo), bez předchozích rodinných případů.
Příznaky
Projev onemocnění je velmi variabilní — od téměř bezpříznakových forem až po těžké, život ohrožující postižení. Mezi běžné známky a symptomy patří:
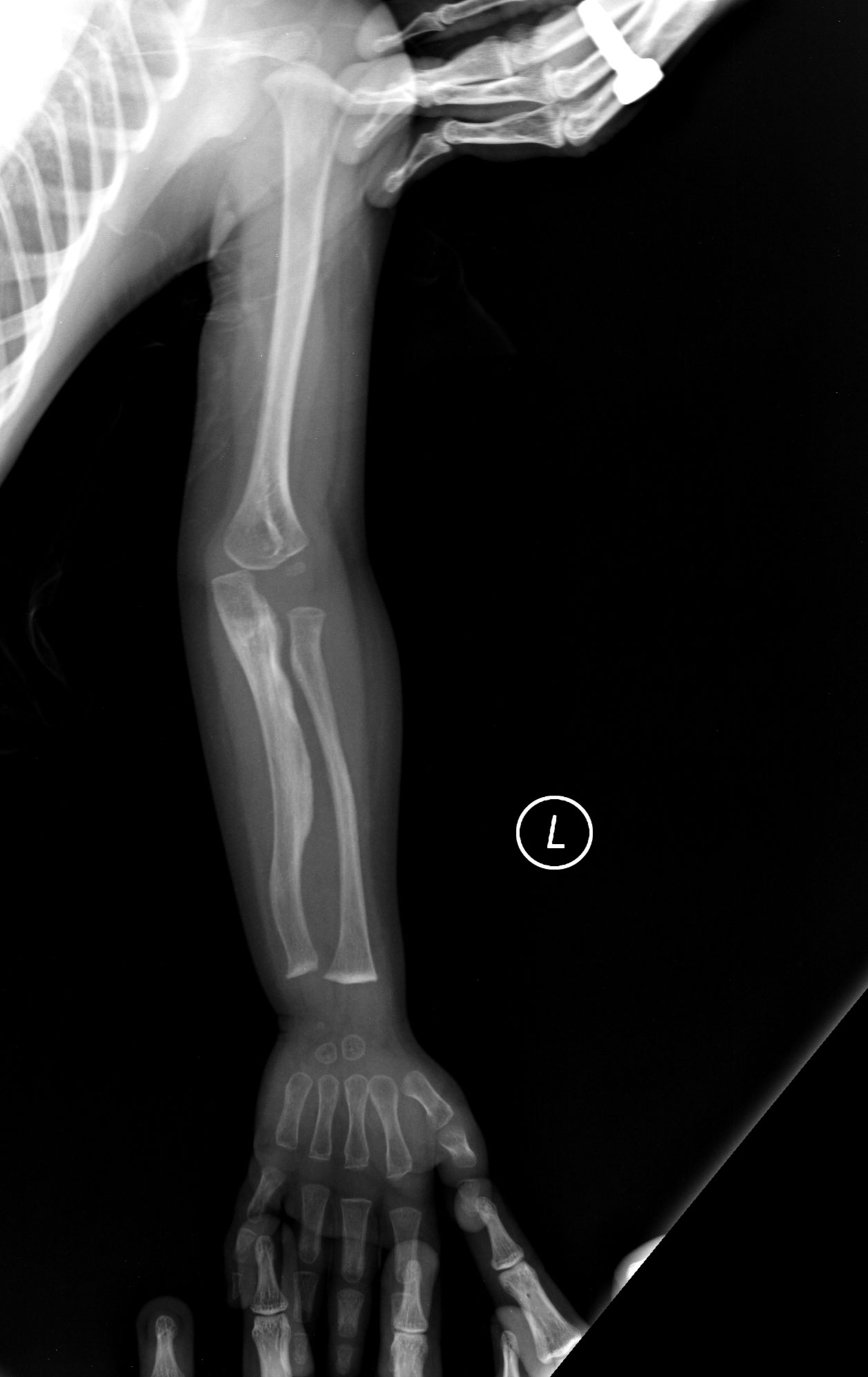
- četné zlomeniny kostí, často po relativně drobných úrazech;
- zkrácený vzrůst a deformity kostry (kyfóza, skolióza, deformace dlouhých kostí);
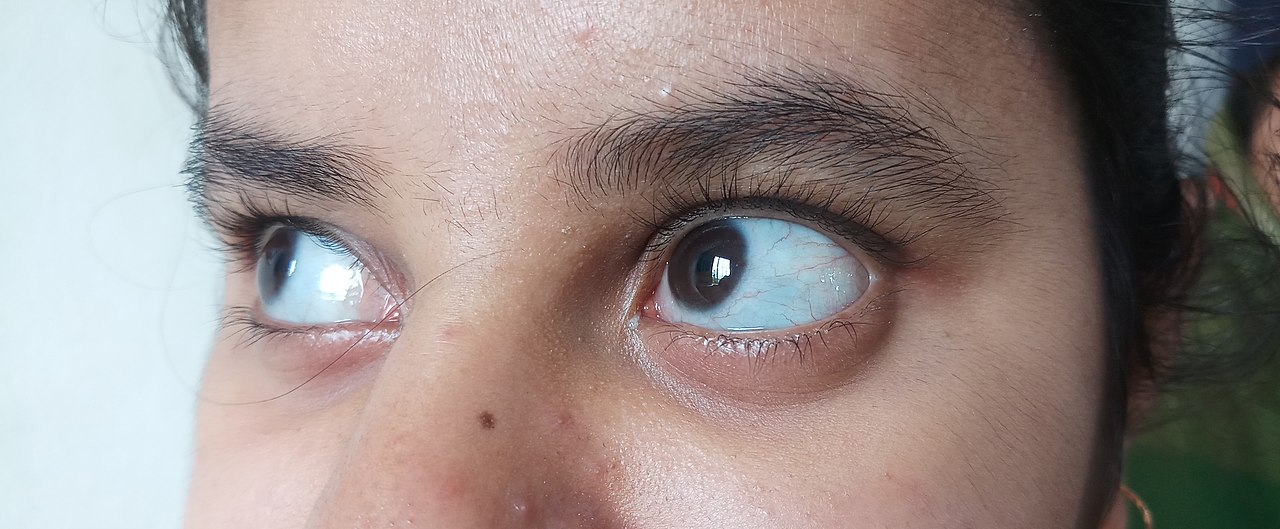
- modré, fialové nebo šedé zbarvení bělma oka (tzv. modré skléry) v důsledku tenké skléry, která propouští barvu cév pod ní;
- dentinogenesis imperfecta — změny na zubech (křehké, matné zuby se sklony k rychlejšímu opotřebení a kazivosti);
- porucha sluchu — u cca 50 % postižených může dojít k progresivní ztrátě sluchu v dospělosti;
- nízká svalová síla, kloubní hypermobilita a vyšší náchylnost k poraněním;
- u těžších forem riziko respiračních komplikací a u novorozenců také zvýšené riziko mortality.
Typy OI
Tradičně se OI dělí podle Sillence na typy I–IV:
- Typ I: nejčastější a nejlehčí forma; časté zlomeniny, modré skléry, normální nebo lehce snížená výška, časté problémy se zuby; inteligence většinou normální.
- Typ II: nejzávažnější forma, často letální v časném novorozeneckém období kvůli mnoha zlomeninám, těžkým deformitám a respiračním problémům.
- Typ III: progresivně těžká forma s vícečetnými zlomeninami, závažnými deformitami kostí, sníženým růstem; modré skléry a často i poruchy sluchu.
- Typ IV: středně těžká forma mezi typy I a III; variabilní průběh, deformity kostí a riziko zlomenin.
Dnes známe i další typy (V a více), které mají specifické rysy (např. radiologické nálezy nebo odlišný dědičný vzorec). Klasifikace se v posledních letech rozšířila na základě genetických nálezů.
Diagnóza a vyšetření
- Klinické vyšetření: rodinná anamnéza, počet a lokalizace zlomenin, vzhled skléry, tvar a kvalita zubů.
- RTG snímky: deformity kostí, snížená kostní denzita, srůstové změny.
- Laboratorní a genetické testy: identifikace mutací v genech COL1A1, COL1A2 a dalších genech. Genetické vyšetření umožňuje potvrdit diagnózu a stanovit dědičný vzorec.
- Další specializovaná vyšetření: audiometrie (sluch), dentální vyšetření, plicní funkční testy u těžších forem.
- Prenatální diagnostika: u rodin s prokázanou mutací je možná prenatální nebo preimplantační genetická diagnostika.
Léčba a péče
OI není v současnosti vyléčitelné, ale existuje řada opatření ke zmírnění příznaků, prevenci komplikací a zlepšení kvality života:
- Ortopedická péče: adekvátní léčba zlomenin, imobilizace, fyzikální terapie, korekční osteotomie a v některých případech vnitřní dlahování či intramedulární dříkování (rodding) k posílení dlouhých kostí.
- Farmakoterapie: bisfosfonáty (např. pamidronát, alendronát, zoledronát) zvyšují kostní denzitu a snižují riziko zlomenin, zejména u dětí. Doplňkově se podává vitamin D a vápník.
- Fyzioterapie a rehabilitace: posilování svalů, nácvik bezpečného pohybu, snížení rizika pádů, udržení mobility a soběstačnosti.
- Stomatologická péče: sledování a léčba dentinogenesis imperfecta, prevence kazů a ochranná ošetření.
- Audiologická péče: pravidelné kontroly sluchu, nasazení sluchadel nebo kochleárních implantátů při potřebě.
- Psychosociální podpora: psychologické poradenství, podpora rodin a sociální služby k zajištění kvalitního života.
Prevence a genetické poradenství
Vzhledem k hereditární povaze onemocnění je důležité genetické poradenství pro rodiny s OI. Možnosti zahrnují testování nosičů, prenatální diagnostiku a preimplantační genetickou diagnostiku (PGD) pro páry, které chtějí snížit riziko přenosu onemocnění na potomky.
Prognóza
Prognóza závisí na typu OI a závažnosti postižení. Mírné formy (typ I) umožňují často relativně normální délku života a soběstačnost, i když s vyšším rizikem zlomenin. Těžké formy (typ II, III) mohou být spojeny s vážnými komplikacemi, zkrácenou délkou života a potřebou intenzivní medicínské péče. Včasná multidisciplinární péče (ortopedie, rehabilitace, genetik, stomatolog, audiologie) výrazně zlepšuje výsledky a kvalitu života pacientů.
Pokud máte podezření na OI u sebe nebo u člena rodiny, je vhodné obrátit se na odborníka (genetika nebo specializované centrum) pro vyšetření, potvrdění diagnózy a plán péče.
Příznaky
Méně závažné příznaky OI mohou zahrnovat:
- snadno zlomené kosti
- uvolněné klouby
- nízký svalový tonus
- modrá, fialová nebo šedá barva normálně bílé části očí.
- trojúhelníkový tvar obličeje
- sklon k rozvoji skoliózy
- křehké zuby
OI má mnoho dalších závažných a smrtelných příznaků, včetně dýchacích problémů a deformací kostí.
Demografické údaje
Otázky a odpovědi
Otázka: Co je to osteogenesis imperfecta?
Odpověď: Osteogenesis imperfecta je genetické onemocnění, kterému se běžně říká křehké kosti. Oslabuje nebo ničí kolagenovou tyčinku, která zajišťuje pevnost kostí, a vede k tomu, že se kosti častěji lámou.
Otázka: Jak se osteogenesis imperfecta dědí?
Odpověď: Osteogenesis imperfecta je obvykle autozomálně dominantní onemocnění, což znamená, že člověk ji může dostat, pokud má abnormální gen pouze jeden z jeho rodičů.
Otázka: Kdo jako první identifikoval osteogenesis imperfecta?
Odpověď: Vrolik poprvé identifikoval osteogenesis imperfecta v roce 1849.
Otázka: Lze osteogenesis imperfecta vyléčit?
Odpověď: Osteogenesis imperfecta se bohužel nedá vyléčit.
Otázka: Jaké jsou čtyři typy osteogenesis imperfecta?
Odpověď: Čtyři typy osteogenesis imperfecta jsou typ jedna, typ dva, typ tři a typ čtyři.
Otázka: Jaké jsou příznaky osteogenesis imperfecta třetího typu?
Odpověď: Lidé s osteogenesis imperfecta třetího typu mohou mít před pubertou více než 100 zlomenin. Jejich oči mají často fialový, modrý nebo šedý odstín a lidé s tímto případem také často ztrácejí sluch.
Otázka: Jak častá je ztráta sluchu u lidí s osteogenesis imperfecta?
Odpověď: U 50 % lidí, kteří mají osteogenesis imperfecta, dochází ke ztrátě sluchu v dospělosti.
Související články
Autor
AlegsaOnline.com Osteogenesis imperfecta – dědičná nemoc křehkých kostí: příčiny, příznaky a typy Leandro Alegsa
URL: https://cs.alegsaonline.com/art/73422
Zdroje
- oif.org : "Osteogenesis Imperfecta Foundation:"
- nlm.nih.gov : "Autosomal dominant: MedlinePlus Medical Encyclopedia"
- oif.org : "Osteogenesis Imperfecta Foundation: Understanding Bone Structure"